|
|
自行写代码进行KEGG富集分析时需要制作富集背景文件,将所有代谢通路中包含的所有基因(蛋白质)或代谢物提取出来,生成背景文件。
KEGGREST:A package that provides a client interface to the Kyoto Encyclopedia of Genes and Genomes (KEGG) REST server.
该软件包为使用者提供了KEGG数据库的API接口,首先简单看一下它的基本使用:
# 查看KEGG包含的子数据库
> listDatabases()
[1] "pathway" "brite" "module" "ko" "genome" "vg" "ag" "compound" "glycan" "reaction"
[11] "rclass" "enzyme" "disease" "drug" "dgroup" "environ" "genes" "ligand" "kegg"
# 获取KEGG数据库中某个物种的所有通路(如人类)
> keggList("pathway","hsa")
# 获取某一条KEGG通路的全部信息,部分结果如图1所示。
> keggGet("hsa00020")
# KEGG通路的基因、代谢物等信息就包含在keggGet()函数获得的结果中

图1 通路hsa00020的部分信息
一、提取某条KEGG通路中的所有基因
# 获取某一条KEGG通路的全部信息
path <- keggGet(&#34;hsa00010&#34;)
head(path)
[[1]]
[[1]]$ENTRY
Pathway
&#34;hsa00010&#34;
……
# 获取通路中的基因信息
gene.info <- path[[1]]$GENE
gene.info
[1] &#34;3101&#34;
[2] &#34;HK3; hexokinase 3 [KO:K00844] [EC:2.7.1.1]&#34;
[3] &#34;3098&#34;
[4] &#34;HK1; hexokinase 1 [KO:K00844] [EC:2.7.1.1]&#34;
……
# 提取gene symbol和Entrez ID
genes <- unlist(lapply(gene.info,function(x) strsplit(x,&#34;;&#34;)))
gene.symbol <- genes[1:length(genes)%%3 == 2]
gene.id <- genes[1:length(genes)%%3 == 1]
gene.symbol
[1] &#34;HK3&#34; &#34;HK1&#34; &#34;HK2&#34; &#34;HKDC1&#34; &#34;GCK&#34; &#34;GPI&#34; &#34;PFKM&#34; &#34;PFKP&#34; &#34;PFKL&#34; &#34;FBP1&#34; &#34;FBP2&#34; &#34;ALDOC&#34; ……
gene.id
[1] &#34;3101&#34; &#34;3098&#34; &#34;3099&#34; &#34;80201&#34; &#34;2645&#34; &#34;2821&#34; &#34;5213&#34; &#34;5214&#34; &#34;5211&#34; &#34;2203&#34; &#34;8789&#34; &#34;230&#34; &#34;226&#34; &#34;229&#34; ……
# 生成gene symbol和Entrez ID匹配的数据框
gene.df <- data.frame(gene.symbol = gene.symbol,gene.id = gene.id)
head(gene.df)
gene.symbol gene.id
1 HK3 3101
2 HK1 3098
3 HK2 3099
# 提取通路ID和通路名称(有需要的自己添加到数据框gene.df中即可)
> path[[1]]$ENTRY
Pathway
&#34;hsa00010&#34;
> path[[1]]$NAME
[1] &#34;Glycolysis / Gluconeogenesis - Homo sapiens (human)&#34;提取某条KEGG通路的基因(名称和ID)时有一个需要注意的点,就是有的基因可能有Entrez ID,但没有gene symbol,此时提取会出错。
二、提取某条KEGG通路中的所有代谢物
# 获取某一条KEGG通路的全部信息
path <- keggGet(&#34;hsa00010&#34;)
# 获取通路中的代谢物信息
compound <- path[[1]]$COMPOUND
compound
C00022
&#34;Pyruvate&#34;
C00024
&#34;Acetyl-CoA&#34;
# 获取代谢物名称和CPD ID
cpd.name <- as.character(compound)
cpd.id <- names(compound)
# 生成代谢物名称和CPD ID匹配的数据框
metabolism.df <- data.frame(cpd.name = cpd.name,cpd.id = cpd.id)
head(metabolism.df)
cpd.name cpd.id
1 Pyruvate C00022
2 Acetyl-CoA C00024
3 D-Glucose C00031
# 如果需要通路的ID和名称,操作同上三、提取所有人类KEGG通路中的所有代谢物
# BiocManager::install(&#34;KEGGREST&#34;,force = TRUE)
# devtools::install_github(&#34;https://github.com/cran/RbioRXN.git&#34;)
# BiocManager::install(&#34;fmcsR&#34;)
library(KEGGREST)
library(fmcsR)
library(plyr)
library(devtools)
library(RbioRXN)
library(stringr)
library(dplyr)
hsa_pathway <- keggList(&#34;pathway&#34;,&#34;hsa&#34;) # 获取KEGG数据库中所有人类通路
hsa_path <- data.frame(hsa_pathway) # 转成数据框,方便后续分析
hsa_path$pathID <- substr(rownames(hsa_path),6,nchar(rownames(hsa_path)[1])) # 提取pathway ID
# 提取通路中的所有代谢物ID及名称
match.df <- vector()
for (i in 1:nrow(hsa_path)) {
hsa_info <- keggGet(hsa_path[i,&#34;pathID&#34;])
hsa_compound <- hsa_info[[1]]$COMPOUND
path_name <- hsa_info[[1]]$NAME
if(length(hsa_compound)>0)
{
cpd <- names(hsa_compound[1])
cpd_name <- as.character(hsa_compound[1])
for (j in 1:(length(hsa_compound)-1)) {
cpd <- paste(cpd,names(hsa_compound)[j+1],sep = &#34;;&#34;)
cpd_name <- paste(cpd_name,as.character(hsa_compound)[j+1],sep = &#34;;&#34;)
}
match.ver <- c(hsa_path[i,&#34;pathID&#34;],path_name,cpd,cpd_name)
match.df <- rbind(match.df,match.ver)
}
if(length(hsa_compound)==0){
match.df <- rbind(match.df,c(hsa_path[i,&#34;pathID&#34;],path_name,&#34;&#34;,&#34;&#34;))
}
}
rownames(match.df) <- match.df[,1]
colnames(match.df) <- c(&#34;Pathway_ID&#34;,&#34;Pathway_Name&#34;,&#34;Compound_ID&#34;,&#34;Metabolism_Name&#34;)
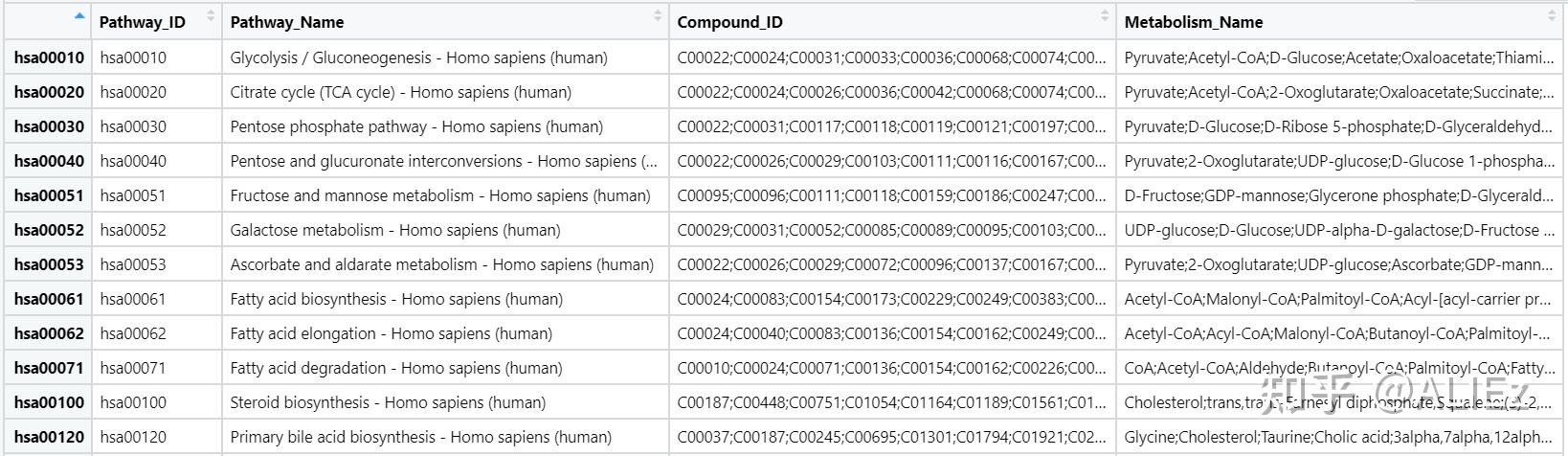
图2 match.df数据框部分截图
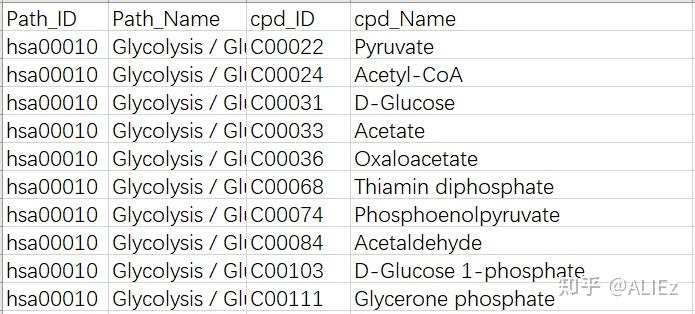
图3 代谢物背景文件
自写代码进行富集分析时,需要制作富集背景文件。“提取所有人类KEGG通路中的所有代谢物”部分的R代码提供了制作代谢物背景文件的一种方法,生成的数据框中,每一行表示一条KEGG通路,获得的代谢物名称和ID分别存储在一列中。如果是需要如图3所示的格式(每一行为一个代谢物),对循环稍作修改即可。
制作基因富集背景文件同理,写循环完成即可。
2022.04.21 Thursday 更新
最近KEGG富集分析的背景基因更新了,我在重新制作背景文件时,发现有更简单的获取KEGG通路包含的所有基因相关信息。KEGGREST包提供了KEGG数据库的API接口,但实际上KEGG数据库网页上也是直接有API的,之前我倒是知道,但使用不熟练,也用的比较少,没想到今天真香了。直接从网站下载数据比写循环来得更简单。
KEGG API:https://www.kegg.jp/kegg/rest/keggapi.html
关于KEGG API的其他使用方法,网站是给出了示例的,感兴趣的可自行研究。
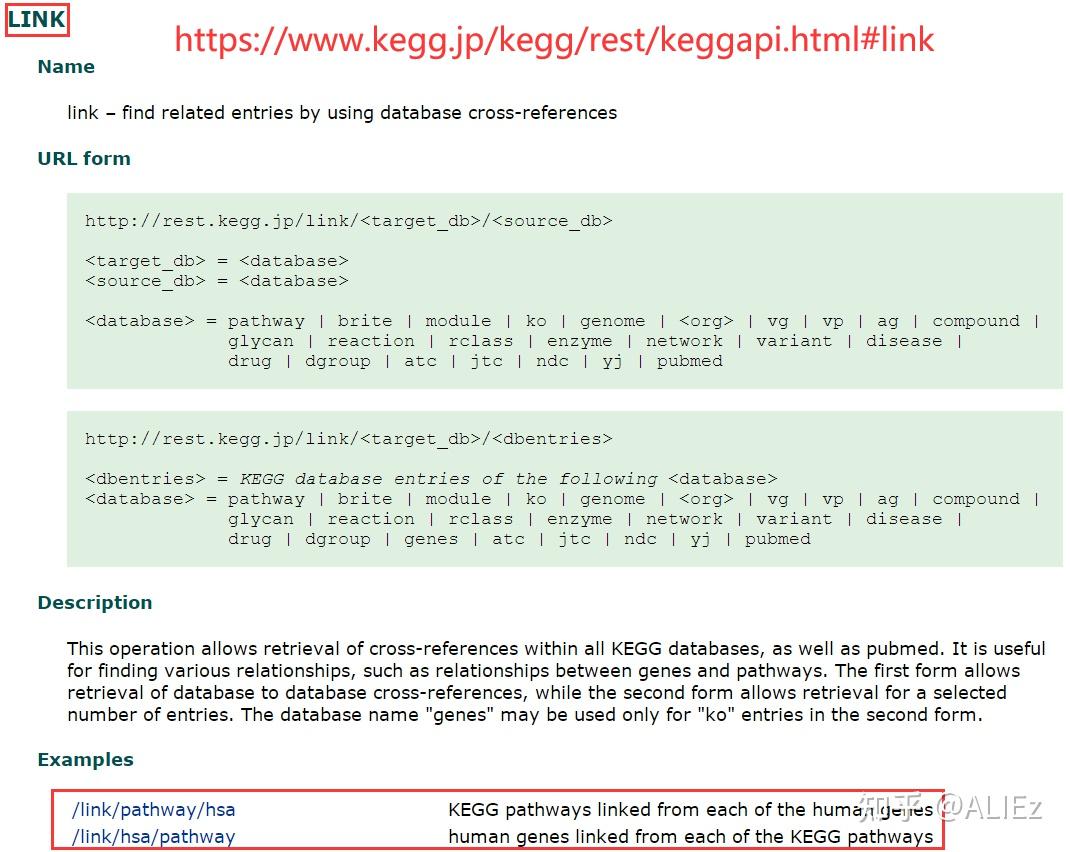
图4 KEGG API提供的人类通路和基因的信息
/link/pathway/hsa和/link/hsa/pathway中提供的信息是相同的,即所有参与人类KEGG通路的基因,选择其中一个下载即可,这两个文件只提供KEGG通路ID和基因ID,如果想获得通路名称,就需要另一个文件。如果不想获取通路名称,那这个文件已经足够用来进行富集分析了。

图5 KEGG API提供的人类通路ID和名称的对应关系
/list/pathway/hsa文件提供了人类KEGG通路ID和名称的对应关系。
有了这两个文件,再使用R语言合并数据不能更简单了。比起使用R包,效率会更高。
2022.12.08 Thursday
回看自己半年前写的代码,这什么玩意儿?!太辣鸡了……
对原本的代码进行了优化,就不再贴出来了。 |
|



 发表于 2023-1-16 17:42:52
发表于 2023-1-16 17:42:52